Chapter 5
Conducting Clinical Research
IN THIS CHAPTER
 Planning and carrying out an experimental clinical trial study
Planning and carrying out an experimental clinical trial study
Protecting the participants
Collecting, validating, and analyzing research data
This chapter provides a closer look at a special kind of human research — the clinical trial. The purpose of a clinical trial is to test one or more interventions, such as a medication or other product or action thought to be therapeutic (such as drinking green tea or exercising). One of the important features of a clinical trial is that it is an experimental study design, meaning that participants in the study are assigned by the study staff which intervention to take. Therefore, there are serious ethical considerations around clinical trials. On the other hand, the clinical trial study design provides the highest quality evidence you can obtain to determine whether or not an intervention actually works, which is a form of causal inference. In this chapter, we cover approaches to designing and executing a high-quality clinical trial and explain the ethical considerations that go along with this.
Designing a Clinical Trial
Clinical trials should conform to the highest standards of scientific rigor, and that starts with the design of the study. The following sections note some aspects of good experimental design.
Identifying aims, objectives, hypotheses, and variables
The aims or goals of a clinical trial are short general statements (often just one statement) of the overall purpose of the experiment. For example, the aim of an experiment may be “to assess whether drinking green tea every day improves alertness in older adults.”
The objectives are much more specific than the aims. In a clinical trial, the objectives usually refer to the effect of the intervention (treatment being tested) on specific outcome variables at specific points in time in a group of a specific type of study participants. In a drug trial, which is a type of experimental clinical research, an efficacy study may have many individual efficacy objectives, as well as one or two safety objectives, while a safety study may or may not have efficacy objectives.
 When designing a clinical trial, you should identify one or two primary objectives — those that are most directly related to the aim of the study. This makes it easier to determine whether the intervention meets the objectives once your analysis is complete. You may then identify up to several dozen secondary objectives, which may involve different variables or the same variables at different time points or in different subsets of the study population. You may also list a set of exploratory objectives, which are less important, but still interesting. Finally, if testing a risky intervention (such as a pharmaceutical), you should list one or more safety objectives (if this is an efficacy study) or some efficacy objectives (if this is a safety study).
When designing a clinical trial, you should identify one or two primary objectives — those that are most directly related to the aim of the study. This makes it easier to determine whether the intervention meets the objectives once your analysis is complete. You may then identify up to several dozen secondary objectives, which may involve different variables or the same variables at different time points or in different subsets of the study population. You may also list a set of exploratory objectives, which are less important, but still interesting. Finally, if testing a risky intervention (such as a pharmaceutical), you should list one or more safety objectives (if this is an efficacy study) or some efficacy objectives (if this is a safety study).
 The objectives you select will determine what data you need to collect, so you have to choose wisely to make sure all data related to those objectives can be collected in the timeframe of your study. Also, these data will be processed from various sources, including case report forms (CRFs), surveys, and centers providing laboratory data. These considerations may limit the objectives you choose to study!
The objectives you select will determine what data you need to collect, so you have to choose wisely to make sure all data related to those objectives can be collected in the timeframe of your study. Also, these data will be processed from various sources, including case report forms (CRFs), surveys, and centers providing laboratory data. These considerations may limit the objectives you choose to study!
Drug clinical trials are usually efficacy studies. Here is an example of each type of objectives you could have in an efficacy study:
- Primary efficacy objective: To compare the effect of new hypertension (HTN) drug XYZ, relative to old drug ABC, on changes in systolic blood pressure (SBP) from baseline to week 12, in participants with HTN.
- Secondary efficacy objective: To compare the effect of HTN drug XYZ, relative to drug ABC, on changes in serum total cholesterol and serum triglycerides from baseline to weeks 4 and 8, in participants with HTN.
- Exploratory efficacy objective: To compare the effect of drug XYZ, relative to drug ABC, on changes in sexual function from baseline to weeks 4, 8, and 12, in male and female subsets of participants with HTN.
- Safety objective: To evaluate the safety of drug XYZ, relative to drug ABC, in terms of the occurrence of adverse events, changes from baseline in vital signs such as temperature and heart rate, and changes in laboratory results of safety panels (including tests on kidney and liver function), in participants with HTN.
For each of these objectives, it is important to specify the time range of participation subject to the analysis (such as the first week of the trial compared to other time segments). Also, which groups are being compared for each objective should be specified.
Hypotheses usually correspond to the objectives but are worded in a way that directly relates to the statistical testing to be performed. So, the preceding primary objective may correspond to the following hypothesis: “The mean 12-week reduction in SBP will be greater in the XYZ group than in the ABC group.” Alternatively, the hypothesis may be expressed in a more formal mathematical notation and as a null and alternate pair (see Chapters 2 and 3 for details on these terms and the mathematical notation used):

where 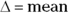 of (
of (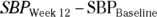 ).
).
In all types of human research, identifying the variables to collect in your study should be straightforward after you’ve selected a study design and enumerated all the objectives. In a clinical trial, you will need to operationalize the measurements you need, meaning you will need to find a way to measure each concept specified in the objectives. Measurements can fall into these categories:
- Administrative: This information includes data related to recruitment, consent, and enrollment, as well as contact information for each participant. You need to keep track of study eligibility documentation, as well as the date of each visit, which study activities took place, and final status at end of study (such as whether participants completed the study, dropped out of the study, or any other outcome).
- Intervention-related: This includes data related to the intervention for each participant, such as group assignment, dosing level, compliance, and adherence measures. If you plan to assign a participant to a particular group but they end up in another group, you need to keep track of both group assignments.
- Outcome-related: This includes ensuring you are measuring both efficacy and safety outcomes on a regular schedule that is documented. You may be asking the participant to keep records, or they may need to be measured in person (to obtain laboratory values, X-rays, and other scans, ECGs, and so on).
- Potential confounding variables: These variables are determined by way of what is known about participants’ relationship with the intervention, outcome, and study eligibility criteria. Typically, potential confounding variables include basic demographic information such as date of birth, gender, and ethnicity. Confounders could also be measured with questions posed to the participant about health behaviors such as tobacco use, exercise patterns, and diet. There are also questions about medical history, including current conditions, past hospitalizations, family medical history, and current and past medication use. Measurements such as height and weight as well as other physical measurements can also be included.
Values that do not change over time, such as birth date and medical history, only need to be recorded at the beginning of the study. In designs including follow-up visits, values that change over time are measured multiple times over the duration of data collection for the study. Depending on the research objectives, these could include weight, medication use, and test results. Most of this data collection is scheduled as part of study visits, and but some may be recorded only at unpredictable times, if at all (such as adverse events, and withdrawing from the study before it is completed).
Deciding who is eligible for the study
Because you can’t examine the entire population for whom the intervention you’re studying is intended, you must select a sample from that population. How you filter in the right sample for your study is by explicitly defining the criteria a potential participant has to meet to be eligible to be enrolled and maintained in the study as a participant.
- Inclusion criteria are used during the screening process to identify potential participants who are members of the population about whom you want to draw conclusions. A reasonable inclusion criterion for a study of a lipid-lowering intervention would be, “Participant must have a documented diagnosis of hyperlipidemia, defined as total cholesterol
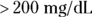 and
and 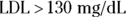 at screening.”
at screening.” - Exclusion criteria are used to identify potential participants who do not fall in the population being studied. They are also used to rule out participation by individuals who are otherwise in the population being studied but should not participate for practical reasons (such as safety, or risk of privacy breach). A reasonable exclusion criterion for a study of a lipid-lowering treatment would be, “Participants who are not willing to change their medication during the study are not eligible to participate.”
- Withdrawal criteria apply to the follow-up portion of the study. They describe situations that could arise during the study that would put the participant in a state where participation should no longer take place. One example would be that the participant is diagnosed with Alzheimer’s disease during the study and can no longer make decisions on their own. A typical withdrawal criterion may be, “If the participant no longer has decision-making capacity, they will be withdrawn.”
Choosing the structure of a clinical trial
Many clinical trials include a comparison of two or more interventions. These types of clinical trials typically have one of the following structures (or designs), each of which has pros and cons:
- Parallel: In this clinical trial design, each participant receives one of the interventions, and the groups are compared. Parallel designs are simpler, quicker, and easier for each participant than crossover designs, but you need more participants for the statistics to work out. Trials with very long treatment periods usually have to be parallel.
- Crossover: In a crossover design, each clinical trial participant receives all the interventions in sequence during consecutive treatment periods (called phases) separated by washout intervals (lasting from several days to several weeks). Crossover designs can be more efficient because each participant serves their own control, eliminating inter-participant variability. But you can use crossover designs only if you’re certain that at the end of each washout period, the participant will have been restored to the same condition as at the start of the study. This may be impossible for studies of progressive diseases, like cancer or emphysema, or for drugs that last a long time in the body and are hard to wash out, like SSRIs and marijuana.
Using randomization
Randomized controlled trials (RCTs) are the gold standard for clinical research (as described in Chapters 7 and 20). In an RCT, the participants are randomly allocated by intervention into treatment groups (in a parallel trial) or into treatment-sequence groups (in a crossover design). Randomization provides several advantages:
- It helps in reducing bias. It specifically helps to eliminate treatment bias, which is where certain treatments are preferentially given to certain participants. A clinician may feel inclined to assign a drug with fewer side effects to healthier participants, but if participants are randomized, then this bias goes away. Another important bias reduced by randomization is confounding, where the treatment groups differ with respect to some characteristic that influences the outcome.
- Randomization makes it easier to interpret the results of statistical testing.
It facilitates blinding. Blinding (also called masking) refers to concealing the identity of the intervention from both participants and researchers. There are two types of blinding:
- Single-blinding: Where participants don’t know what intervention they’re receiving, but the researchers do.
- Double-blinding: Where neither the participants nor the researchers know which participants are receiving which interventions.
- Note: In all cases of blinding, for safety reasons, it is possible to unblind individual participants, as at least one of the members of the research team has the authority to unblind.
Blinding eliminates bias resulting from the placebo effect, which is where participants tend to respond favorably to any treatment (even a placebo), especially when the efficacy variables are subjective, such as pain level. Double-blinding also eliminates deliberate and subconscious bias in the investigator’s evaluation of a participant’s condition.
The simplest kind of randomization involves assigning each newly enrolled participant to a treatment group by the flip of a coin or a similar method. But simple randomization may produce an unbalanced pattern, like the one shown in Figure 5-1 for a small study of 12 participants and two treatments: Drug (D) and Placebo (P).

© John Wiley & Sons, Inc.
FIGURE 5-1: Simple randomization.
If you were hoping to have six participants in each group, you won’t be pleased if you end up with three participants receiving the drug and nine receiving the placebo, because it’s unbalanced. But unbalanced patterns like this arise quite often from 12 coin flips. (Try it if you don’t believe us.) A better approach is to require six participants in each group but shuffle those six Ds and six Ps around randomly, as shown in Figure 5-2.

© John Wiley & Sons, Inc.
FIGURE 5-2: Random shuffling.
This arrangement is better because there are exactly six participants assigned to drug and placebo each. But this particular random shuffle happens to assign more drugs to the earlier participants and more placebos to the later participant (which is just by chance). If the recruitment period is short, this would be perfectly fine. However, if these 12 participants were enrolled over a period of five or six months, seasonal effects may be mistaken for treatment effects, which is an example of confounding.
To make sure that both treatments are evenly spread across the entire recruitment period, you can use blocked randomization, in which you divide your subjects into consecutive blocks and shuffle the assignments within each block. Often the block size is set to twice the number of treatment groups. For instance, a two-group study would use a block size of four. This is shown in Figure 5-3.

© John Wiley & Sons, Inc.
FIGURE 5-3: Blocked randomization.
You can create simple and blocked randomization lists in Microsoft Excel using the RAND() built-in function to shuffle the assignments. You can also use the web page at https://www.graphpad.com/quickcalcs/randomize1.cfm to generate blocked randomization lists quickly and easily.
Selecting the analyses to use
You should select the appropriate analytic approach for each of your study hypotheses based on the type of data involved, the structure of the study, and the requirements of the hypothesis. The rest of this book describes statistical methods to analyze the kinds of data you’re likely to encounter in human research. Your strategy is to apply them to a clinical trial design. In clinical trials, changes in values of variables over time, and differences between treatments in crossover studies are often analyzed by paired t tests and repeated-measures ANOVAs.
Differences between groups of participants in parallel studies are often analyzed by unpaired t tests and ANOVAs. Often, final regression models are developed for clinical trial interpretation because these can control for residual confounding (which are covered in the chapters in Part 5). In longer clinical trials, time until death (survival time) and the times to the occurrence of other endpoint events (besides death) are analyzed by survival methods (Part 6 focuses on survival analysis methods).
Determining how many participants to enroll in a clinical trial
Chapter 3 presents the concept of statistical power, and for a clinical trial, you should enroll enough participants to provide sufficient statistical power when testing the primary objective of the study. The specific way you calculate the required sample size depends on the statistical test that’s used for the primary hypothesis. Each chapter of this book that describes hypothesis tests also shows how to estimate the required sample size for that test. To get quick sample-size estimates, you can use G*Power (an application for sample-size calculations described in Chapter 4), or you can use the formulas, tables, and charts in Chapter 25 and on the book’s Cheat Sheet at www.dummies.com (just search for “Biostatistics For Dummies Cheat Sheet”).
You must also allow some extra space in your target sample-size estimate for some of the enrolled participants to drop out or otherwise not contribute the data you need for your analysis. For example, suppose that you need full data from 64 participants for sufficient statistical power to answer your main objective. If you expect a 15 percent attrition rate from the study, which means you expect only 85 percent of the enrolled participants to have analyzable data, then you need to plan to enroll 64/0.84, or 76, participants in the study.
Assembling the study protocol
A study protocol (or just protocol) is a document that lays out exactly what you plan to do to collect and analyze data in a research study. For ethical reasons, every research study involving human participants should have a protocol, and for other types of studies, having a protocol prepared before starting the research is considered best practices. In a clinical trial, the protocol is especially important because the participants are being assigned to interventions by the researcher, and there is often double-blinding and randomization.
In terms of standard elements, a formal drug clinical trial protocol typically contains these components:
Title: A title conveys as much information about the trial as you can fit into one sentence, including the protocol ID, name of the study, clinical phase, type and structure of trial, type of randomization and blinding, name of the drug or drugs being tested, treatment regimen, intended effect, and the population being studied (which could include a reference to individuals with a particular medical condition). A title can be quite long — this example title has all the preceding elements:
Protocol BCAM521-13-01 (ASPIRE-2) — a Phase-IIa, double-blind, placebo-controlled, randomized, parallel-group study of the safety and efficacy of three different doses of AM521, given intravenously, once per month for six months, for the relief of chronic pain, in adults with knee osteoporosis.
- Background information: This section includes information about the disease for which the drug is an intended treatment. It includes the epidemiology (the condition’s prevalence and impact), and its known physiology down to the molecular level. It also includes a review of treatments currently available (if any), and information about the drug or drugs being tested, including mechanism of action, the results of prior testing, and known and potential risks and benefits to participants.
- Rationale: The rationale for the study states why it makes sense to do this study at this time and includes a justification for the choice of doses, how the drug is administered (such as orally or intravenously), duration of drug administration, and follow-up period.
- Aims, objectives, and hypotheses: We discuss these items in the earlier section “Identifying aims, objectives, hypotheses, and variables.”
- Detailed descriptions of all inclusion, exclusion, and withdrawal criteria: See the earlier section “Deciding who is eligible for the study” for more about these terms.
- Design of the clinical trial: As described in the earlier section “Choosing the structure of a clinical trial,” the clinical trial’s design defines its structure. This includes the number of treatment groups as well as consecutive stages of the study. These stages could include eligibility screening, washout, treatment, follow-up, and so on. This section often includes a schematic diagram of the structure of the study.
- Drug description: This description details each drug that will be administered to the participants. This includes the chemical composition with the results of chemical analysis of the drug, if available. It also includes instructions about how to store, prepare, and administer the drug correctly.
- Blinding and randomization schemes: These schemes include descriptions of how and when the study will be unblinded. This includes the emergency unblinding of individual participants if necessary. See the earlier section “Using randomization” for more information.
- Procedural descriptions: This section describes every procedure that will be performed at every visit. These include administrative procedures, such as enrollment and informed consent, and diagnostic procedures, such as physical exams and measuring vital signs. It covers all activities where data are collected from participants in the study.
- Safety considerations: These factors include the known and potential side effects of each drug included. This section also includes the known and potential side effects of each procedure in the study, including X-rays, MRI scans, and blood draws. It also describes steps taken to minimize the risk to the participants.
- Handling of adverse events: This section describes how adverse events will be addressed should they occur during the study. It includes a description of the data that will be recorded, including the nature of the adverse event, severity, dates and times of onset and resolution, any medical treatment given for the event, and whether or not the investigator thinks the event was related to the study drug. It also explains how the research study will support the participant after the adverse event.
- Definition of safety, efficacy, and other analytical populations: This section includes definitions of safety and efficacy variables and endpoints. In other words, this section defines variables or changes in variables that serve as indicators of safety or efficacy.
- Planned enrollment and analyzable sample size: Justification for these numbers must also be provided.
- Proposed statistical analyses: Some protocols describe, in detail, every analysis for every objective. Others include only a summary and refer to a separate Statistical Analysis Plan (SAP) document for details of the proposed analysis. This section should also include descriptions of how missing data will be handled analytically, adjustments for multiple testing to control Type I errors (see Chapter 3), and whether any interim analyses are planned. If a separate SAP is used, it typically contains a detailed description of all the calculations and analyses that will be carried out on the data, including the descriptive summaries of all data and the testing of all the hypotheses specified in the protocol. The SAP also usually contains mock-ups called shells of all the tables, listings, and figures (referred to as TLFs) that will be generated from the data.
It will also contain administrative details, like names and contact information for the research team, a table of contents, a list of abbreviations, description of policies of data handling, and financing and insurance agreements.
Carrying Out a Clinical Trial
After you’ve designed your clinical trial and have described it in the protocol document, it’s time to move on to the next step. The operational details will, of course, vary from one study to another, but a few aspects apply to all clinical trials. In any study involving human participants, the most important consideration is protecting those participants from harm.
Since the end of World War II, international agreements have established ethical guidelines for human research all over the world. Regardless of the country in which you are conducting research, prior to beginning a human research study, your protocol will need to be approved by at least one ethics board. Selection of ethics boards for human research depends upon where the research is taking place and what institutions are involved (see the later section “Working with Institutional Review Boards”).
Protecting clinical trial participants
In any research involving humans, two issues are of utmost importance:
- Safety: Minimizing the risk of physical harm to the participant from the drug or drugs being tested and from the procedures involved in the study
- Privacy/confidentiality: Ensuring that data collected during the study are not breached (stolen) and are not made public in a way that identifies a specific participant without the participant’s consent
The following sections describe some of the infrastructure that helps protect human subjects.
Surveying regulatory agencies
In the United States, several government organizations oversee human subjects’ protection:
- Commercial pharmaceutical research is governed by the Food and Drug Administration (FDA).
- Most academic biological research is sponsored by the National Institutes of Health (NIH) and is governed by the Office for Human Research Protections (OHRP).
 Because research ethics are international, other countries have similar agencies, so international clinical trial oversight can get confusing. An organization called the International Conference on Harmonization (ICH) works to establish a set of consistent standards that can be applied worldwide. The FDA and NIH have adopted many ICH standards (with some modifications).
Because research ethics are international, other countries have similar agencies, so international clinical trial oversight can get confusing. An organization called the International Conference on Harmonization (ICH) works to establish a set of consistent standards that can be applied worldwide. The FDA and NIH have adopted many ICH standards (with some modifications).
Working with Institutional Review Boards
For all protocols that describe research that could potentially be considered human research, in the United States, an ethics board called an Institutional Review Board (IRB) must review the protocol and approve it (or find it exempt from human research laws) before you can start the research. You have to submit an application along with the protocol and your plans for gaining informed consent of potential participants to an IRB with jurisdiction over your research and ensure that you are approved to proceed before you start.
Most medical centers and academic institutions (as well as some industry partners) run their own IRBs with jurisdiction over research conducted at their institution. If you’re not affiliated with one of these centers or institutions (for example, if you’re a freelance biostatistician or clinician), you may need the services of a consulting or free-standing IRB. The sponsor of the research may recommend or insist you use a particular IRB for the project.
Getting informed consent
An important part of protecting human participants is making sure that they’re aware of the risks of a study before agreeing to participate in it. They also need to be fully informed as to what will happen in the study before they can give consent to participate. You must prepare an Informed Consent Form (ICF) describing, in simple language, the nature of the study, why it is being conducted, what is being tested, what procedures participants will undergo, and the risks and benefits of participation. The ICF is used to guide study staff when they explain the study to potential participants, who are then expected to sign it if they want to participate in the study.
Potential participants must be told that they can refuse to participate with no penalty to them, and if they join the study, they can withdraw at any time for any reason without fear of retribution or the withholding of regular medical care. The IRB can provide ICF templates with examples of their recommended or required wording.
Prior to performing any procedures on a potential participant (including screening tests involved in determining eligibility), study staff must go through an approved ICF document with the potential participant and give them time to read it and decide whether or not they want to participate. If they choose to participate, the ICF must be signed and witnessed. The signed ICFs must be retained as part of the official documentation for the project, along with all data collected (including laboratory reports, ECG tracings, and records of all test products administered to the participants), as well as records of all procedures performed on participants. The sponsor, the regulatory agencies, the IRB, and other entities may ask to review these documents at any time as part of oversight.
Considering data safety monitoring boards and committees
For clinical trials of interventions that are likely to be of low risk (such as drinking green tea), investigators are usually responsible for ensuring participant safety by tracking unexpected adverse events, abnormal laboratory tests, and other red flags during the course of the study. But for studies involving high-risk treatments (like cancer chemotherapy trials), a separate data safety monitoring board or committee (DSMB or DSMC) should be arranged. A DSMB is typically required by the sponsor, the investigator, the IRB, or a regulatory agency. A DSMB typically has about six members, including expert clinicians in the relevant area of research and a statistician, who are external to the study staff. A DSMB meets at regular intervals during the clinical trial to review the unblinded safety data acquired up to that point. The committee is authorized to modify, suspend, or even terminate a study if it has serious concerns about the safety of the participants.
Getting certified in human subjects protection
 As you may have surmised from the preceding sections, clinical research involves regulatory requirements that include penalties for noncompliance. Therefore, you shouldn’t try to guess how to write a research protocol, obtain IRB approval, or engage in any other research methods with which you are unfamiliar, and just hope that everything goes well. You should ensure that you, along with any others who may be assisting you, are properly trained in matters relating to human subjects protection.
As you may have surmised from the preceding sections, clinical research involves regulatory requirements that include penalties for noncompliance. Therefore, you shouldn’t try to guess how to write a research protocol, obtain IRB approval, or engage in any other research methods with which you are unfamiliar, and just hope that everything goes well. You should ensure that you, along with any others who may be assisting you, are properly trained in matters relating to human subjects protection.
Fortunately, such training is readily available. Most hospitals and medical centers provide training in the form of online courses, workshops, lectures, and other resources. As you comply with ongoing IRB training, you receive a certification in human subjects protection. Most IRBs and funding agencies require proof of certification from study staff. If you don’t have access to that training at your institution, you can get certified by taking an online tutorial offered by the NIH (https://grants.nih.gov/policy/humansubjects/research/training-and-resources.htm).
Collecting and validating data
Data in a clinical trial are typically collected digitally and manually. Examples of data collected digitally include a participant filling out an online survey or a blood pressure monitor collecting data from a participant. Data are collected manually when they are written down on paper first, then undergo data entry to become digital. Either way, some paper forms may be included, and many digital forms are created for data entry in a clinical trial. These forms are for data entry of data collected from various parts of the study, but in clinical trial lingo, they are all referred to as case report forms, or CRFs.
- In the case of digitally collected data, the central analytic team will run routines for validating the data. They will communicate with study staff if they find errors and work them out.
- In the case of manually collected data, data entry into a digital format will be required. Study staff typically are expected to log into an online database with CRFs and do data entry from data collected on paper.
- The sponsor of the study will provide detailed data entry instructions and training to ensure high-quality data collection and validation of the data collected in the study.
Analyzing Your Data
In the following sections, we describe some general situations that come up in all clinical research, regardless of what kind of analysis you use.
Dealing with missing data
Most clinical trials have incomplete data for one or more variables, which can be a real headache when analyzing your data. The statistical aspects of missing data are quite complicated, so you should consult a statistician if you have more than just occasional, isolated missing values. Here we describe some commonly used approaches for coping with missing data:
- Exclusion: Exclude a case from an analysis if any of the required variables for that analysis is missing. This seems simple, but the downside to this approach is it can reduce the number of analyzable cases, sometimes quite severely. And if the result is missing for a reason that’s related to treatment efficacy, excluding the case can bias your results.
- Imputation: Imputation is where you replace a missing value with a value you impute, or create yourself. When analysts impute in a clinical trial, they typically take the mean or median of all the available values for that variable and fill that in for the missing variable. In reality, you have to keep the original variable, and then save a separate, imputed variable so that you can document the type of imputation applied. There are a lot of downsides to imputation. If you are imputing a small number of values, it’s not worth it because it adds bias. You may as well just exclude those cases. But if you impute a large number of values, you are basically making up the data yourself, adding more bias.
- Last Observation Carried Forward (LOCF): LOCF is a special case of imputation. Sometimes during follow-up, one of a series of sequential measurements on a particular participant is missing. For example, imagine that there were supposed to be four weekly glucose values measured, and you were missing a measurement only on week three. In that case, you could use the most recent previous value in the series, which is the week two measurement, to impute the week three measurement. This technique is called Last Observation Carried Forward (LOCF) and is one of the most widely used strategies. Although imputation adds bias, LOCF adds bias in the conservative direction, making it more difficult to demonstrate efficacy. This approach is popular with regulators, who want to put the burden of proof on the drug and study sponsor.
Handling multiplicity
Every time you perform a statistical significance test, you run a chance of being fooled by random fluctuations into thinking that some real effect is present in your data when, in fact, none exists (review Chapter 3 for a refresher on statistical testing). If you declare the results of the test are statistically significant, and in reality they are not, you are committing Type I error. When you say that you require p < 0.05 to declare statistical significance, you’re testing at the 0.05 (or 5 percent) alpha (α) level. This is another way of saying that you want to limit your Type I error rate to 5 percent. But that 5 percent error rate applies to each and every statistical test you run. The more analyses you perform on a data set, the more your overall α level increases. If you perform two tests at α = 0.05, your chance of at least one of them coming out falsely significant is about 10 percent. If you run 40 tests, the overall α level jumps to 87 percent! This is referred to as the problem of multiplicity, or as Type I error inflation.
Chapter 11 covers dealing with multiplicity when making multiple comparisons. One approach discussed in Chapter 11 is performing post-hoc tests following an ANOVA for comparing several groups. Post-hoc tests incorporate a built-in adjustment to keep the overall α at only 5 percent across all comparisons. This can be especially important when conducting an interim analysis, or an analysis done before the official end of study data collection. But when you’re testing different hypotheses — like when comparing different variables at different time points between different groups — you are faced with some difficult decisions to make about reducing Type I error inflation.
In sponsored clinical trials, the sponsor and DSMB will weigh in on how they want to see Type I error inflation controlled. If you are working on a clinical trial without a sponsor, you should consult with another professional with experience in developing clinical trial analyses to advise you on how to control your Type I error inflation given the context of your study.
Each time an interim analysis is conducted, a process called data close-out must occur. This creates a data snapshot, and the last data snapshot from a data close-out process produces the final analytic dataset, or dataset to be used in all analyses. Data close-out refers to the process where current data being collected are copied into a research environment, and this copy is edited to prepare it for analysis. These edits could include adding imputations, unblinding, or creating other variables needed for analysis. The analytic dataset prepared for each interim analysis and for final analysis should be stored with documentation, as decisions about stopping or adjusting the trial are made based on the results of interim analyses.